- 医薬品の適正使用・安全対策
- 3.医薬品の安全対策
- 医薬品の安全対策
- Sec.1
1医薬品の安全対策
■医薬品の安全対策
現在、医薬品の市販後の安全対策として、副作用等の情報を収集する制度、収集された安全性情報を評価し適切な措置を講じる体制が整備されているところである。また、医薬品を適正に使用したにもかかわらず生じた健康被害に対する救済制度等が設けられている。これらは、これまでの薬害事件が和解により集結した後、その経験や教訓を踏まえて、拡充されてきたものである。Ⅳ(薬害の歴史)を参照して作成のこと。
■医薬品の副作用情報等の収集、評価及び措置
1961年に起こったサリドマイド薬害事件を契機として、医薬品の安全性に関する問題を世界共通のものとして取り上げる気運が高まり、1968年、世界保健機関(WHO)加盟各国を中心に、各国自らが医薬品の副作用情報を収集、評価する体制(WHO国際医薬品モニタリング制度)を確立することにつながった。
1)副作用情報等の収集
【医薬品・医療機器等安全性情報報告制度】 法第68条の10第2項の規定により、薬局開設者、病院、診療所若しくは飼育動物診療施設の開設者又は医師、歯科医師、薬剤師、登録販売者、獣医師その他の医薬関係者は、医薬品の副作用等によるものと疑われる健康被害の発生を知った場合において、保健衛生上の危害の発生又は拡大を防止するため必要があると認めるときは、その旨を厚生労働大臣に報告しなければならないとされている。なお、実務上は、法第68条の13第3項の規定により、報告書を総合機構に提出することとされている。
本制度は、医薬品の使用、販売等に携わり、副作用等が疑われる事例に直接に接する医薬関係者からの情報を広く収集することによって、医薬品の安全対策のより着実な実施を図ることを目的としており、WHO加盟国の一員としてわが国が対応した安全対策に係る制度の一つである。
本制度は、1967年3月より、約3000の医療機関をモニター施設に指定して、厚生省(当時)が直接副作用報告を受ける「医薬品副作用モニター制度」としてスタートした。また、一般用医薬品による副作用等の情報を収集するため、1978年8月より、約3000のモニター薬局で把握した副作用事例等について、定期的に報告が行われるようになった。その後、1997年7月に「医薬品等安全性情報報告制度」として拡充し、2002年7月には薬事法が改正され、医師や薬剤師等の医薬関係者による副作用等の報告を義務化することにより、副作用等に関する情報の収集体制がより一層強化された。2006年6月の薬事法改正よる登録販売者制度の導入に伴い、登録販売者も本制度に基づく報告を行う医薬関係者として位置づけられている。
【企業からの副作用等の報告制度】 医薬品の市販後においても、常にその品質、有効性及び安全性に関する情報を収集し、また、医薬関係者に必要な情報を提供することが、医薬品の適切な使用を確保する観点からも、企業責任として重要なことである。
製造販売業者等には、法第68条の10第1項の規定に基づき、その製造販売をし、又は承認を受けた医薬品について、その副作用等によるものと疑われる健康被害の発生、その使用によるものと疑われる感染症の発生等を知ったときは、その旨を定められた期限までに厚生労働大臣に報告することが義務づけられている(別表5-4)。なお、実務上は、法第68条の13第3項の規定により、報告書を総合機構に提出することとされている。
なお、薬局開設者、医療施設の開設者、医薬品の販売業者又は医師、歯科医師、薬剤師その他の医薬関係者(登録販売者を含む。)においては、法第68条の2第2項により、製造販売業者等が行う情報収集に協力するよう努めなければならないこととされている。
本制度は、1979年の薬事法改正により制度化され、製造販売業者等に対して国への報告を求めてきたが、その後1996年の薬事法改正により、製造販売業者等が副作用等の情報収集の義務を負うことが明記されている。
1979年に創設された副作用・感染症報告制度において、医薬品等との関連が否定できない感染症に関する症例情報の報告や研究論文等について、製造販売業者等に対して国への報告義務を課しているが、それに加えて2003年7月からは、その前年に行われた薬事法改正により、血液製剤等の生物由来製品を製造販売する企業に対して、当該製品又は当該製品の原料又は材料による感染症に関する最新の論文や知見に基づき、当該企業が製造販売する生物由来製品の安全性について評価し、その成果を定期的に国へ報告する制度を導入している。
一般用医薬品に関しても、承認後の調査が製造販売業者等に求められており、副作用等の発現状況等の収集・評価を通じて、承認後の安全対策につなげている。具体的には既存の医薬品と明らかに異なる有効成分が配合されたものについては、10年を超えない範囲で厚生労働大臣が承認時に定める一定期間(概ね8年)、承認後の使用成績等を製造販売業者等が集積し、厚生労働省へ提出する制度(再審査制度)が適用される。また、医療用医薬品で使用されていた有効成分を一般用医薬品で初めて配合したものについては、承認条件として承認後の一定期間(概ね3年)、安全性に関する調査及び調査結果の報告が求められている。要指導医薬品は、上記と同様に調査結果の報告が求められている。
2)副作用情報等の評価及び措置
収集された副作用等の情報は、その医薬品の製造販売業者等において評価・検討され、必要な安全対策が図られる。各制度により集められた副作用情報については、総合機構において専門委員の意見を聴きながら調査検討が行われ、その結果に基づき、厚生労働大臣は、薬事・食品衛生審議会の意見を聴いて、使用上の注意の改訂の指示等を通じた注意喚起のための情報提供や、効能・効果や用法・用量の一部変更、調査・実験の実施の指示、製造・販売の中止、製品の回収等の安全対策上必要な行政措置を講じている。
【健康危機管理体制の整備】 1997年に厚生省(当時)は、血液製剤によるHIV感染被害を深く反省し、国民の信頼を回復するためには、健康危機管理体制を抜本的に見直すことが必要であるとの認識に立ち、健康危機管理、すなわち、医薬品、食中毒、感染症、飲料水等に起因する、国民の生命、健康の安全を脅かす事態に対して、健康被害の発生予防、拡大防止等の対策を迅速に講じていくための体制を整備した。
健康危機管理に当たっては、国民の生命・健康に関わるという危機意識を常に持ち、事実に対しては予断を持って判断することなく真摯に受け止め、科学的・客観的な評価を行うとともに、情報の広範な収集、分析の徹底と対応方針の弾力的な見直しに努め、国民に対して情報の速やかな提供と公表を行うことを基本としている。
■医薬品による副作用等が疑われる場合の報告の仕方
法第68条の10第2項の規定に基づく医薬品の副作用等報告では、保健衛生上の危害の発生又は拡大を防止するためとの趣旨に鑑みて、医薬品等によるものと疑われる、身体の変調・不調、日常生活に支障を来す程度の健康被害(死亡を含む。)について報告が求められている。なお、医薬品との因果関係が必ずしも明確でない場合であっても報告の対象となり得る。また、安全対策上必要があると認めるときは、医薬品の過量使用や誤用等によるものと思われる健康被害についても報告がなされる必要がある。
医薬品の副作用は、使用上の注意に記載されているものだけとは限らず、また、副作用の症状がその医薬品の適応症状と見分けがつきにくい場合(例えば、かぜ薬による間質性肺炎など)もある。したがって、医薬品の販売等に従事する専門家においては、購入者等からの訴えに素直に耳を傾け、あるいはそのような副作用があるのでないかという、真摯な対応がなされることが重要である。
報告様式(別表5-5)は、医薬品・医療機器等安全性情報と同様、総合機構ホームページから入手できる。
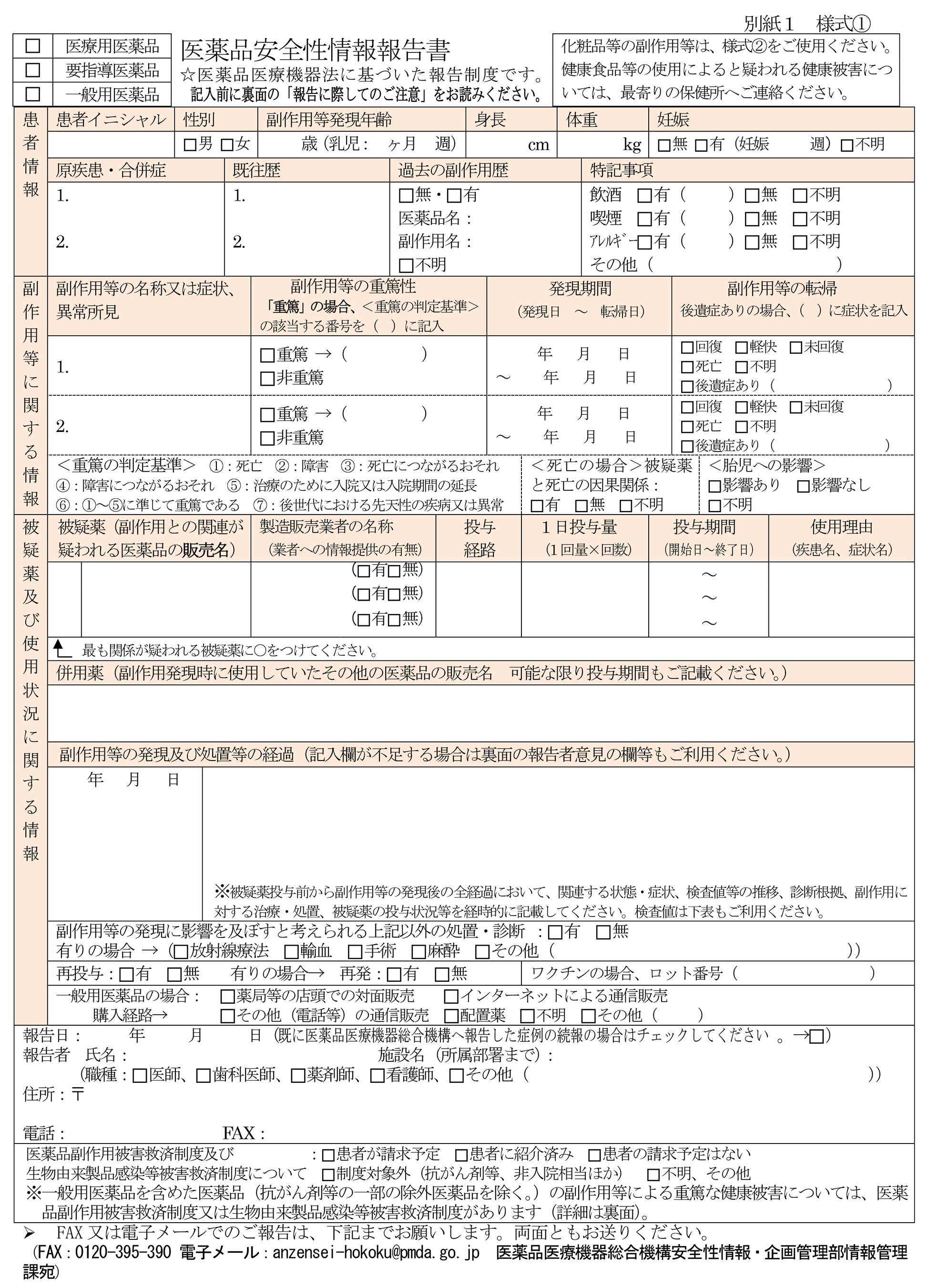

また、関係機関・関係団体の協力の下、医学・薬学関係の専門誌等にも掲載されている。報告様式の記入欄すべてに記入がなされる必要はなく、医薬品の販売等に従事する専門家においては、購入者等(健康被害を生じた本人に限らない)から把握可能な範囲で報告がなされればよい。なお、複数の専門家が医薬品の販売等に携わっている場合であっても、当該薬局又は医薬品の販売業において販売等された医薬品の副作用等によると疑われる健康被害の情報に直接接した専門家1名から報告書が提出されれば十分である。
報告期限は特に定められていないが、保健衛生上の危害の発生又は拡大防止の観点から、報告の必要性を認めた場合においては、適宜速やかに、郵送、ファクシミリ又は電子メールにより、法第68条の13第3項の規定に基づき、報告書(別表5-5)を総合機構に送付することとされている。報告者に対しては、安全性情報受領確認書が交付される。